Publications
Group Publications
Faraday Discussions
· 2026
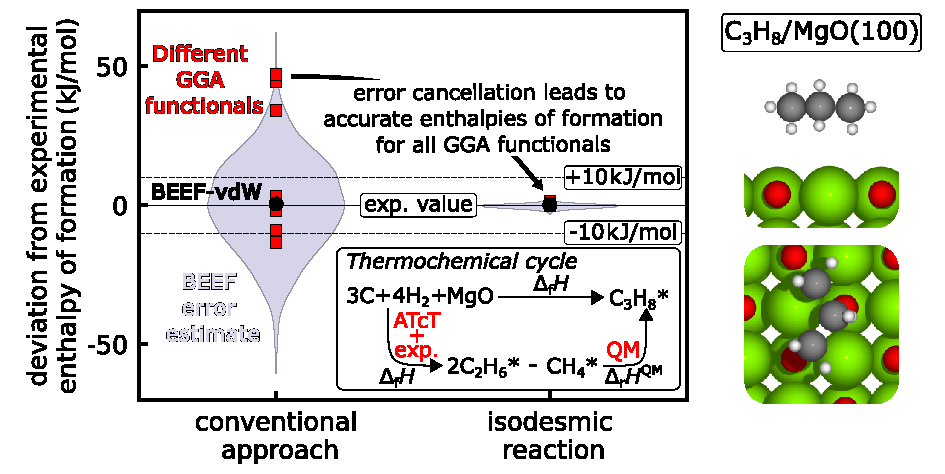
Predictive multiscale modeling of heterogeneously catalyzed reactions requires accurate enthalpies of adsorbates. These properties are typically calculated from density functional theory (DFT) using exchange-correlation functionals with the generalized-gradient approximation (GGA) since more accurate electronic structure methods are not feasible. Therefore, the derived enthalpies are subject to large inaccuracies. We address this challenge through an error-cancellation approach that builds on the connectivity-based hierarchy (CBH) to derive enthalpies of formation of adsorbates with beyond-DFT accuracy without increasing computational cost. This method constructs reactions that conserve the electronic configuration between the target and the reference species, leading to error cancellation. The method is applied to adsorbates on Pt(111), Ni(111), and MgO(100). With the CBH method, it is possible to determine enthalpies of formation that are in excellent agreement with experimental measurements for a range of adsorbates and across many GGA exchange-correlation functionals, clearly outperforming conventional referencing approaches. Additionally, the method combines available experimental surface science data with gas-phase thermochemistry data and DFT data in a global thermochemical network. More accurate enthalpies of formation have a tremendous impact on the predictive performance of multiscale models and enable more conclusive insights into reaction mechanisms of catalytic reactions.

Chemical Society Reviews
· 2025
Thermophysical properties of adsorbates and gas-phase species define the free energy landscape of heterogeneously catalyzed processes and are pivotal for an atomistic understanding of the catalyst performance. These thermophysical properties, such as the free energy or the enthalpy, are typically derived from density functional theory (DFT) calculations. Enthalpies are species-interdependent properties that are only meaningful when referenced to other species. The widespread use of DFT has led to a proliferation of new energetic data in the literature and databases. However, there is a lack of consistency in how DFT data is referenced and how the associated enthalpies or free energies are stored and reported, leading to challenges in reproducing or utilizing the results of prior work. Additionally, DFT suffers from exchange–correlation errors that often require corrections to align the data with other global thermochemical networks, which are not always clearly documented or explained. In this review, we introduce a set of consistent terminology and definitions, review existing approaches, and unify the techniques using the framework of linear algebra. This set of terminology and tools facilitates the correction and alignment of energies between different data formats and sources, promoting the sharing and reuse of ab initio data. Standardization of thermochemistry concepts in computational heterogeneous catalysis reduces computational cost and enhances fundamental understanding of catalytic processes, which will accelerate the computational design of optimally performing catalysts.
Journal of Catalysis
· 2025
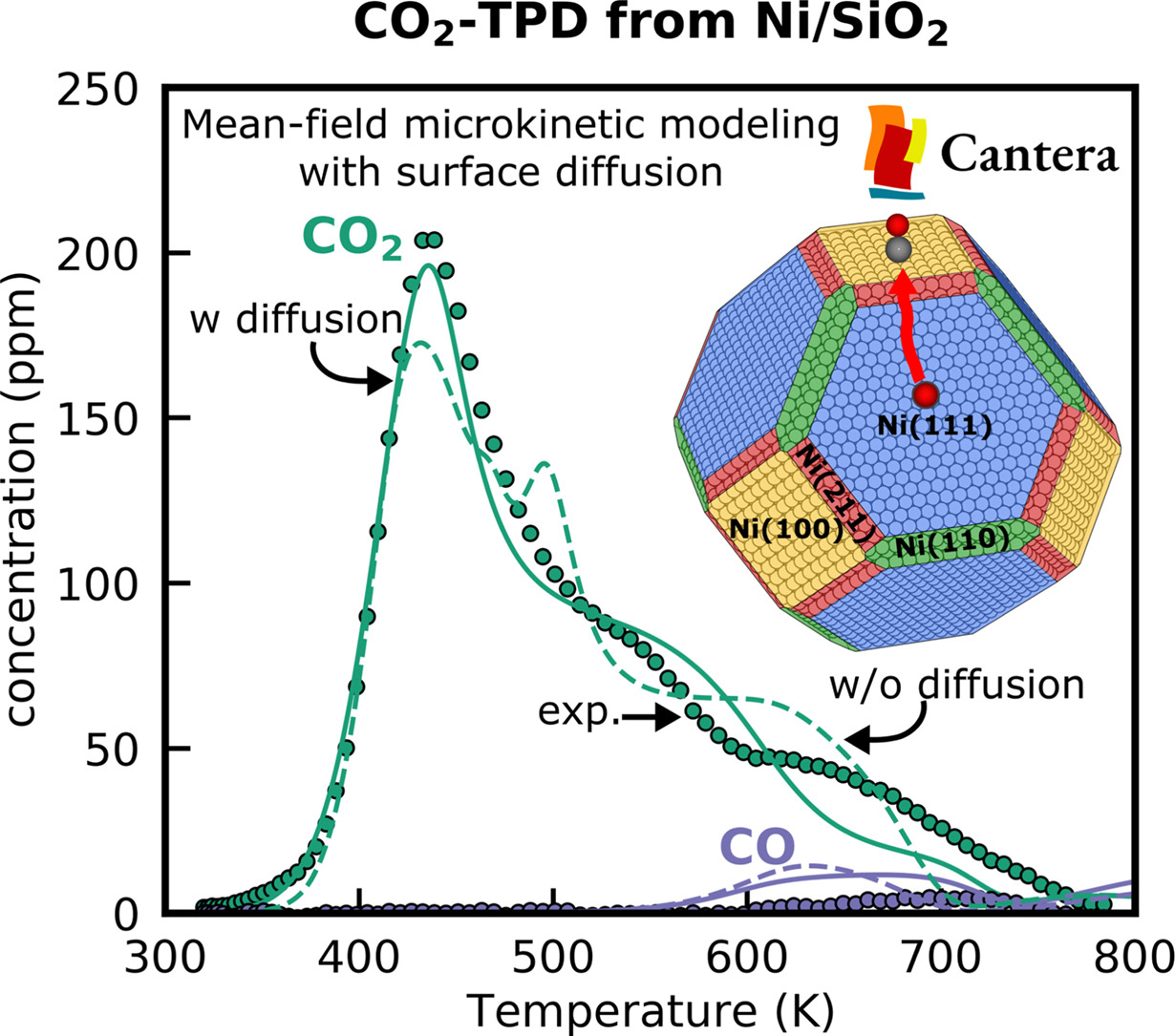
The interaction of CO2 with Ni catalysts is important for many industrial processes like methanation, which is known to be structure sensitive. Consequently, structure-dependent multiscale modeling is required to accurately capture the interaction of CO2 with the various Ni facets and to provide accurate atomistic insights. While mean-field multiscale models can be constructed for multifaceted nanoparticles, surface diffusion of adsorbates between the facets is often not considered. In this study, we close the gap by extending the open-source Cantera toolkit with a universal framework for surface diffusion between facets in mean-field microkinetic models, making it the first widely adopted software tool that includes these features. We leverage these updates to develop a thermodynamically consistent microkinetic model for a Ni nanoparticle consisting of Ni(111), Ni(100), Ni(211), and Ni(110) using data from DFT calculations and single-crystal experiments to unravel the interaction of CO2 with these facets through the simulation of temperature-programmed desorption profiles. Including surface diffusion and coverage effects into the mean-field microkinetic model leads to a significantly improved agreement between experiments from a catalyst and the simulations. Through rigorous correlated uncertainty quantification of all structural and energetic parameters, we are able to identify a microkinetic model within the uncertainty space that is in excellent agreement with the recorded desorption profile. This model highlights that Ni(110), which contributes only to a small extent to the overall Ni surface area, dominates the desorption pattern and that surface diffusion plays a crucial role. The Cantera implementation is generic and can be applied to other metal nanoparticles and metal/metal oxide interfaces, providing a step towards closing the material gap.
All Publications
Faraday Discussions
· 2026
Predictive multiscale modeling of heterogeneously catalyzed reactions requires accurate enthalpies of adsorbates. These properties are typically calculated from density functional theory (DFT) using exchange-correlation functionals with the generalized-gradient approximation (GGA) since more accurate electronic structure methods are not feasible. Therefore, the derived enthalpies are subject to large inaccuracies. We address this challenge through an error-cancellation approach that builds on the connectivity-based hierarchy (CBH) to derive enthalpies of formation of adsorbates with beyond-DFT accuracy without increasing computational cost. This method constructs reactions that conserve the electronic configuration between the target and the reference species, leading to error cancellation. The method is applied to adsorbates on Pt(111), Ni(111), and MgO(100). With the CBH method, it is possible to determine enthalpies of formation that are in excellent agreement with experimental measurements for a range of adsorbates and across many GGA exchange-correlation functionals, clearly outperforming conventional referencing approaches. Additionally, the method combines available experimental surface science data with gas-phase thermochemistry data and DFT data in a global thermochemical network. More accurate enthalpies of formation have a tremendous impact on the predictive performance of multiscale models and enable more conclusive insights into reaction mechanisms of catalytic reactions.
Current Opinion in Chemical Engineering
· 2026
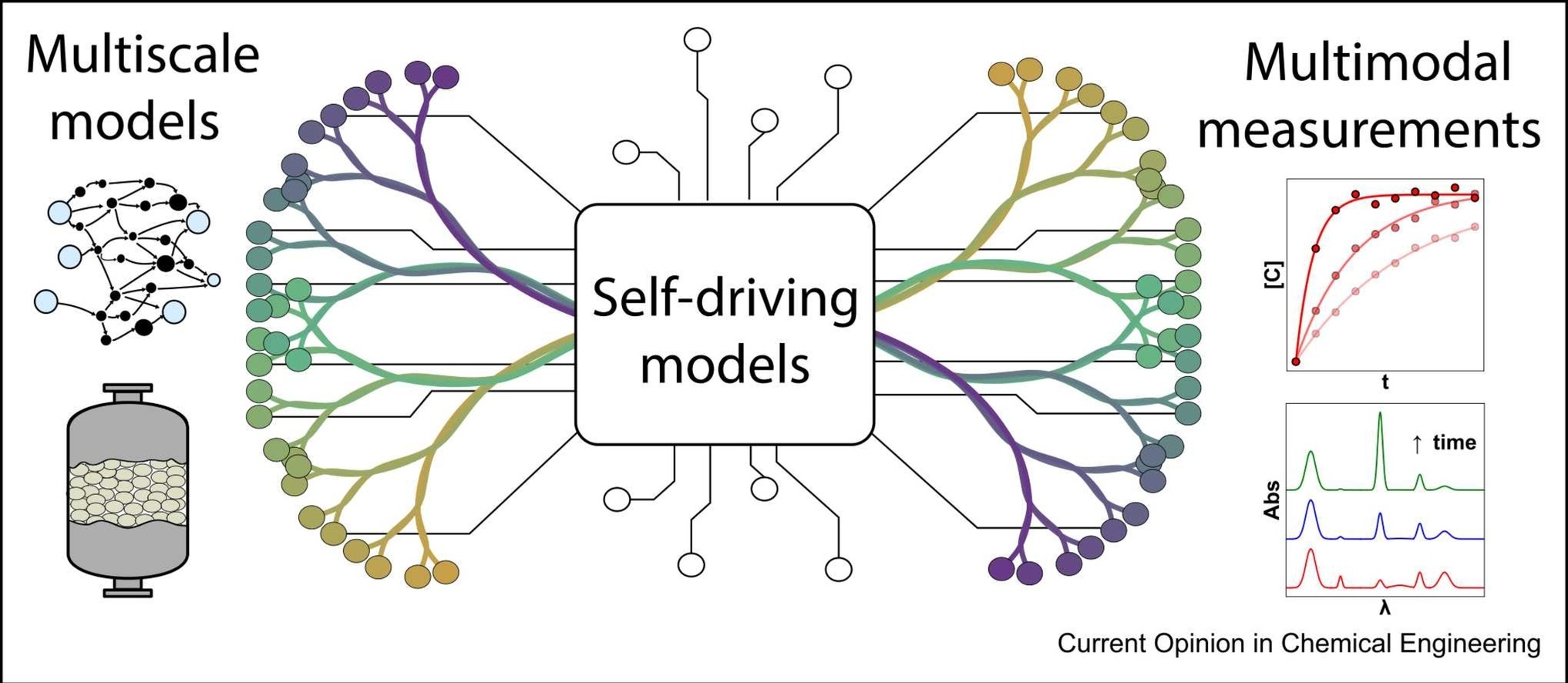
Artificial intelligence (AI) is influencing heterogeneous catalysis research by accelerating simulations and materials discovery. A key frontier is integrating AI with multiscale models and multimodal experiments to address the many-to-one challenge of linking intrinsic kinetics to observables. Advances in machine-learned force fields, microkinetics, and reactor modeling enable rapid exploration of chemical spaces, while operando and transient data provide unprecedented insight. Yet, inconsistent data quality and model complexity limit mechanistic discovery. Generative and agentic AI can automate model generation, quantify uncertainty, and couple theory with experiment, realizing self-driving models that produce interpretable, reproducible, and transferable understanding of catalytic systems.

Chemie Ingenieur Technik
· 2025
The Nachwuchs Reaktionstechnik (NaWuReT) is an organization of early-career chemical engineers in the DECHEMA/VDI subject division Reaction Engineering. In the spring of 2024, the annual colloquium was held as a series of online lectures featuring five distinguished late-career academics and experts from industry. The speakers shared insights into how they transformed their efforts and ideas into successful careers in academia and industry. Additionally, they provided valuable recommendations for early-career chemical engineers, offering a critical resource for professional development in the field.
Computers & Chemical Engineering
· 2025
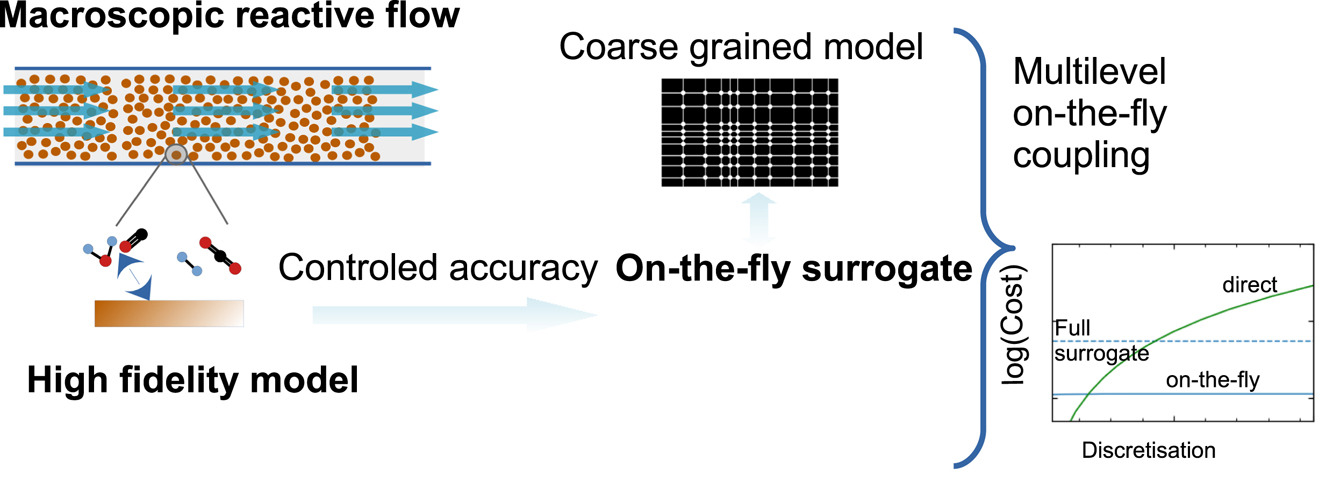
Coupling microscopic high-fidelity models, such as microkinetic models, into continuum scale simulations can easily become intractable in practice due to the costs of the high-fidelity model evaluation. To lift this burden, we present a novel multilevel self-consistent on-the-fly sparse grid approach, which integrates the construction of surrogates of the high-fidelity model in a multilevel fashion into the continuum solution process. Besides its efficiency, an appealing feature of the approach is its simplicity and robustness. A single hyperparameter controls the whole workflow, from training set design to the accuracy of the reactor model. We demonstrate the methodology on a recent microkinetic model for catalytic combustion in a fixed-bed reactor model as a representative example. Already with modest numbers of data, the approach achieves sufficient accuracy, reducing the effort by orders of magnitude compared to a direct coupling.
Chemical Society Reviews
· 2025
Thermophysical properties of adsorbates and gas-phase species define the free energy landscape of heterogeneously catalyzed processes and are pivotal for an atomistic understanding of the catalyst performance. These thermophysical properties, such as the free energy or the enthalpy, are typically derived from density functional theory (DFT) calculations. Enthalpies are species-interdependent properties that are only meaningful when referenced to other species. The widespread use of DFT has led to a proliferation of new energetic data in the literature and databases. However, there is a lack of consistency in how DFT data is referenced and how the associated enthalpies or free energies are stored and reported, leading to challenges in reproducing or utilizing the results of prior work. Additionally, DFT suffers from exchange–correlation errors that often require corrections to align the data with other global thermochemical networks, which are not always clearly documented or explained. In this review, we introduce a set of consistent terminology and definitions, review existing approaches, and unify the techniques using the framework of linear algebra. This set of terminology and tools facilitates the correction and alignment of energies between different data formats and sources, promoting the sharing and reuse of ab initio data. Standardization of thermochemistry concepts in computational heterogeneous catalysis reduces computational cost and enhances fundamental understanding of catalytic processes, which will accelerate the computational design of optimally performing catalysts.
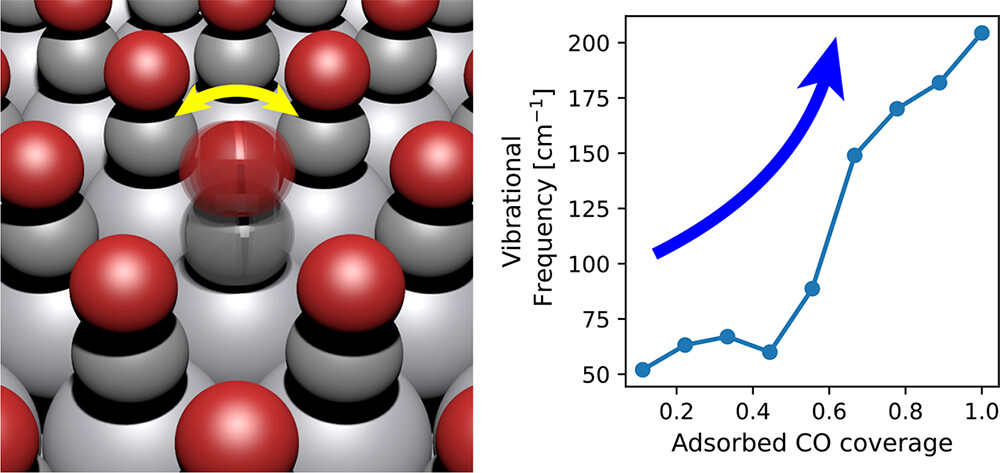
Journal of Chemical Information and Modeling
· 2025
This work focuses on the impact of lateral interactions on the thermophysical properties of adsorbates. We present different parametrizations for coverage-dependent enthalpy, entropy, and heat capacity in a mean-field microkinetic model. These models are tested against two systems, CO/Pt(111) and CO/Co(0001), using two different functionals. A detailed investigation into how coverage influences the thermophysical properties of CO* is presented. We place particular emphasis on studying the impact of coverage on the vibrational partition function and how this affects the entropy of adsorbates. Higher coverages typically lead to increased repulsive interactions, which should further constrain the large amplitude modes that contribute the most to the vibrational entropy. In some cases, however, the opposite effect occurred; the vibrational entropy actually increased because surface crowding forced adsorbates to different binding locations that had lower frequencies. Our results highlighted cases where coverage-dependent entropy should be included, such as for adsorbates with lateral vibrational modes and systems at high temperatures. These methods for including coverage-dependent properties into mean-field microkinetics in a thermodynamically consistent way are now available in the open-source software Cantera.
Journal of Catalysis
· 2025
The interaction of CO2 with Ni catalysts is important for many industrial processes like methanation, which is known to be structure sensitive. Consequently, structure-dependent multiscale modeling is required to accurately capture the interaction of CO2 with the various Ni facets and to provide accurate atomistic insights. While mean-field multiscale models can be constructed for multifaceted nanoparticles, surface diffusion of adsorbates between the facets is often not considered. In this study, we close the gap by extending the open-source Cantera toolkit with a universal framework for surface diffusion between facets in mean-field microkinetic models, making it the first widely adopted software tool that includes these features. We leverage these updates to develop a thermodynamically consistent microkinetic model for a Ni nanoparticle consisting of Ni(111), Ni(100), Ni(211), and Ni(110) using data from DFT calculations and single-crystal experiments to unravel the interaction of CO2 with these facets through the simulation of temperature-programmed desorption profiles. Including surface diffusion and coverage effects into the mean-field microkinetic model leads to a significantly improved agreement between experiments from a catalyst and the simulations. Through rigorous correlated uncertainty quantification of all structural and energetic parameters, we are able to identify a microkinetic model within the uncertainty space that is in excellent agreement with the recorded desorption profile. This model highlights that Ni(110), which contributes only to a small extent to the overall Ni surface area, dominates the desorption pattern and that surface diffusion plays a crucial role. The Cantera implementation is generic and can be applied to other metal nanoparticles and metal/metal oxide interfaces, providing a step towards closing the material gap.

Chemie Ingenieur Technik
· 2024
The Nachwuchs Reaktionstechnik (NaWuReT) is an organization of early-career chemical engineers of the DECHEMA/VDI subject division Reaction Engineering. In the spring of 2023, we organized a series of online lectures involving five invited speakers from industry to shed light on different career opportunities. The speakers gave insights into their individual career pathways and presented their current projects, both of which sparked active discussions among speakers and attendees.
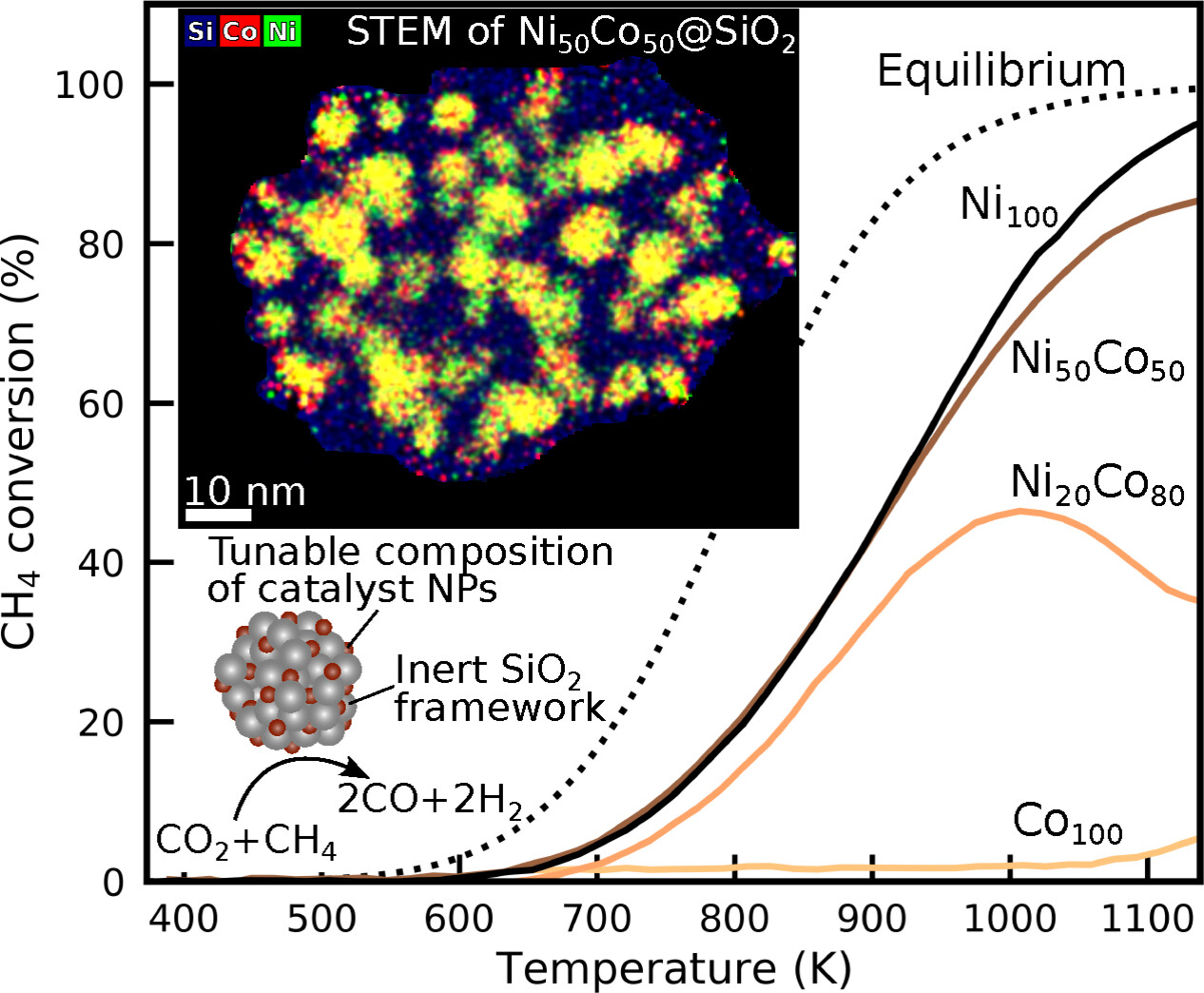
ChemCatChem
· 2024
Dry reforming of methane (DRM) is an attractive reaction for synthesis gas production, since it converts two greenhouse gases into valuable chemical feedstock. Silica supported bimetallic catalysts with constant metal loading but varying Ni/Co ratios (xNi=1, 0.8, 0.6, 0.5, 0.4, 0.2, 0) were produced via spray-drying for the application in DRM, aiming at the identification of compositions with superior activity. In this study, building block particles refer to an inert framework, supporting the catalyst. The elemental distribution of Si, Ni, and Co within such building block particles was evaluated using scanning transmission electron microscopy. Additionally, X-ray diffraction and temperature-programmed reduction experiments confirmed that Ni and Co initially exist in an oxidic state within the SiO2 framework after production and later transform into an alloyed metallic state upon reduction with hydrogen, as confirmed by X-ray photoelectron spectroscopy. A systematic comparison of the activity over a temperature range (323–1150 K) was performed using temperature scanning measurements. The highest intrinsic activity was found with the bimetallic Ni40Co60 particles.
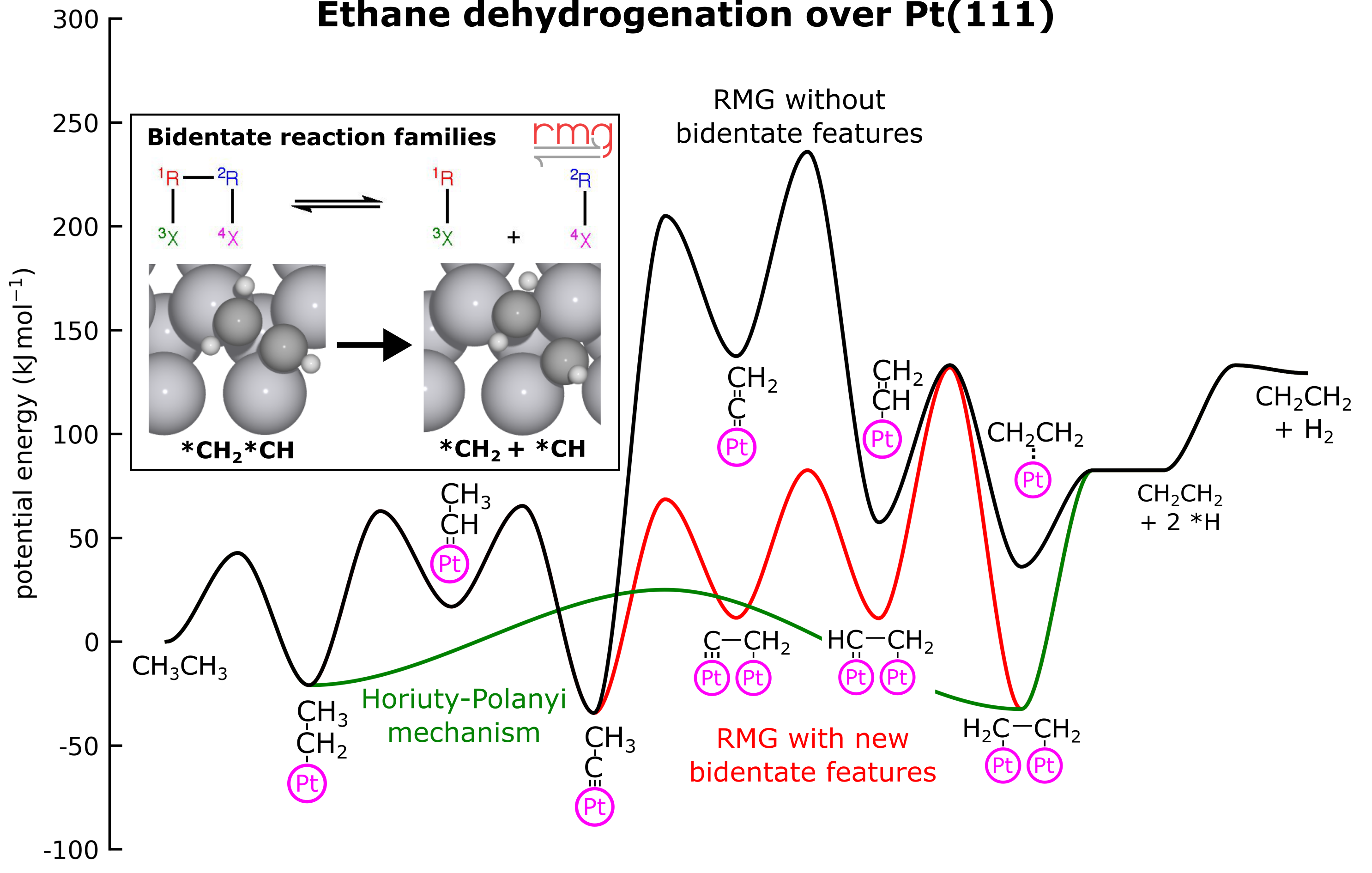
Digital Discovery
· 2024
The open-source Reaction Mechanism Generator (RMG) has been enhanced with new features to handle multidentate adsorbates. New reaction families have been added based upon ab initio data from 26 reactions involving CxOyHz bidentate adsorbates with two heavy atoms on Pt(111). Additionally, the estimation routines for thermophysical properties were improved and extended towards bidentate species. Non-oxidative dehydrogenation of ethane over Pt(111) is used as a case study to demonstrate the effectiveness of these new features. RMG not only discovered the pathways from prior literature but also uncovered new elementary steps involving abstraction reactions. Various mono- and bimetallic catalysts for this process were screened using linear scaling relations within RMG, where a unique mechanism is generated for each catalyst. These results are consistent with prior literature trends, but they add additional insight into the rate-determining steps across the periodic table. With these additions, RMG can now explore more intricate reaction mechanisms of heterogeneously catalyzed processes for the conversion of larger molecules, which will be particularly important in fuel synthesis.
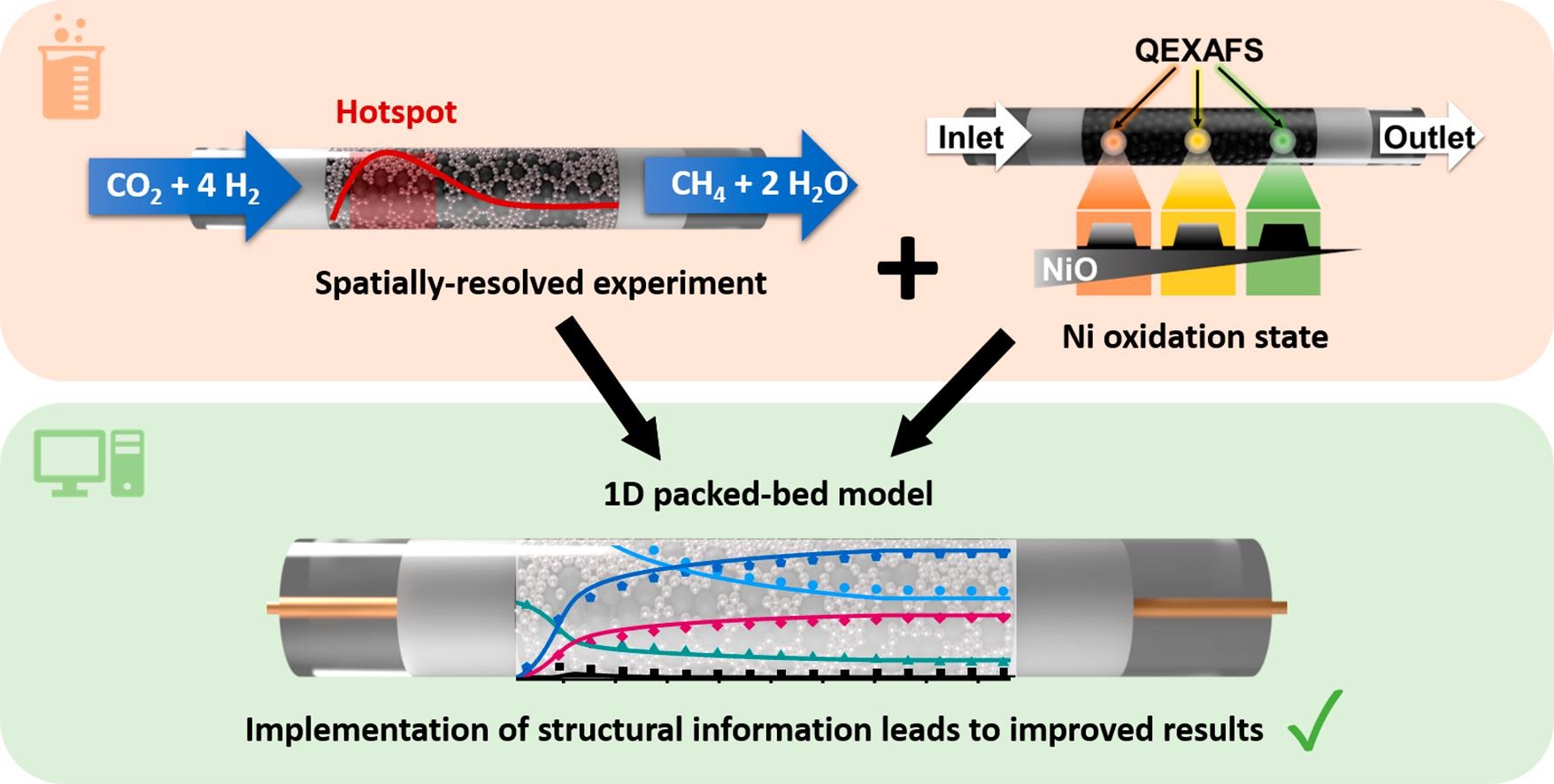
Chemical Engineering Journal
· 2023
CO2 methanation via the Sabatier reaction with (green) H2 is promising due to its role in achieving a carbon–neutral energy balance in the context of Power-to-Gas technologies. Since Ni-based catalysts are relatively inexpensive compared to other metals and exhibit high catalytic activity, they are most commonly used. Due to the exothermic nature of the reaction, strong temperature and concentration gradients occur, which influence the catalyst structure. Thus, revealing the effects of structural changes of the catalyst along the reactor bed on local activity and selectivity is essential. A 1D packed-bed reactor model was used for numerical simulations, coupled with detailed microkinetics and mass transport limitations. The simulation results are compared with axially-resolved concentration and temperature profiles over 17 wt% Ni/γ-Al2O3 and 17 wt% Ni3.2Fe/γ-Al2O3 catalysts at oven temperatures of 623 K and 723 K. Using additional information from structural spatially-resolved synchrotron-based operando X-ray absorption spectroscopy studies, the oxidation state of Ni was considered in modeling the reactor by changing the catalytically active surface area along the reactor. Predicted surface coverages are compared with surface species experimentally determined by diffuse reflectance infrared Fourier transform spectroscopy. Overall, this study demonstrates the importance of combining modeling with spatially-resolved and temperature-dependent experiments to improve multiscale models and make predictions more accurate.
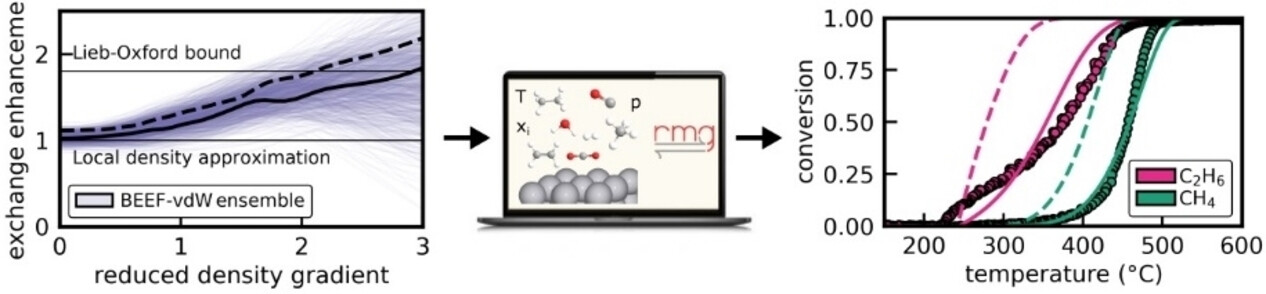
Angewandte Chemie International Edition
· 2023
The study presents an ab-initio based framework for the automated construction of microkinetic mechanisms considering correlated uncertainties in all energetic parameters and estimation routines. 2000 unique microkinetic models were generated within the uncertainty space of the BEEF-vdW functional for the oxidation reactions of representative exhaust gas emissions from stoichiometric combustion engines over Pt(111) and compared to experiments through multiscale modeling. The ensemble of simulations stresses the importance of considering uncertainties. Within this set of first-principles-based models, it is possible to identify a microkinetic mechanism that agrees with experimental data. This mechanism can be traced back to a single exchange-correlation functional, and it suggests that Pt(111) could be the active site for the oxidation of light hydrocarbons. The study provides a universal framework for the automated construction of reaction mechanisms with correlated uncertainty quantification, enabling a DFT-constrained microkinetic model optimization for other heterogeneously catalyzed systems.
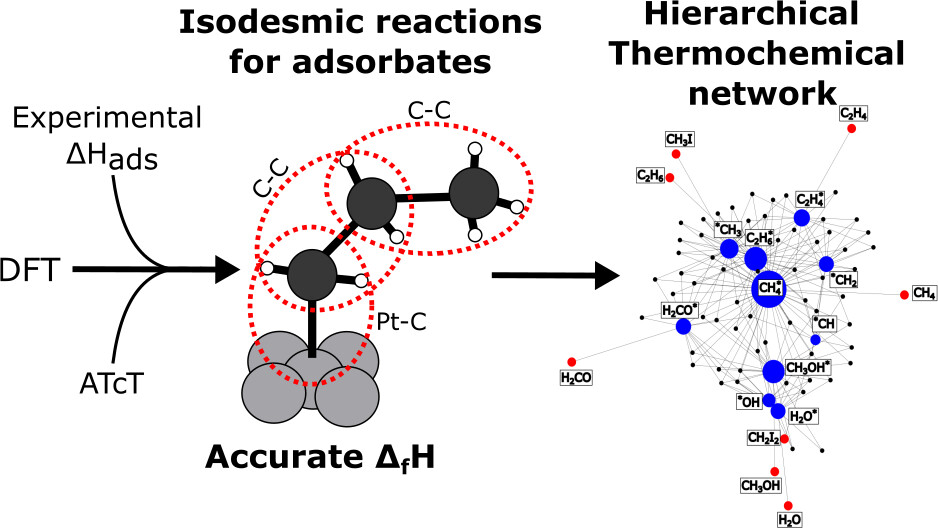
Journal of Chemical Theory and Computation
· 2023
Enthalpies of formation of adsorbates are crucial parameters in the microkinetic modeling of heterogeneously catalyzed reactions since they quantify the stability of intermediates on the catalyst surface. This quantity is often computed using density functional theory (DFT), as more accurate methods are computationally still too expensive, which means that the derived enthalpies have a large uncertainty. In this study, we propose a new error cancellation method to compute the enthalpies of formation of adsorbates from DFT more accurately through a generalized connectivity-based hierarchy. The enthalpy of formation is determined through a hypothetical reaction that preserves atomistic and bonding environments. The method is applied to a data set of 60 adsorbates on Pt(111) with up to 4 heavy (non-hydrogen) atoms. Enthalpies of formation of the fragments required for the bond balancing reactions are based on experimental heats of adsorption for Pt(111). The comparison of enthalpies of formation derived from different DFT functionals using the isodesmic reactions shows that the effect of the functional is significantly reduced due to the error cancellation. Thus, the proposed methodology creates an interconnected thermochemical network of adsorbates that combines experimental with ab initio thermochemistry in a single and more accurate thermophysical database.

Chemie Ingenieur Technik
· 2022
The Nachwuchs Reaktionstechnik (NaWuReT) are early-career scientists from the ProcessNet Division Reaction Engineering. In autumn 2021, they organized an online colloquium with international early-career scientists from the chemical engineering community. Five guests were invited to give a scientific talk and provide insights into their career paths. The guests gave advice and emphasized the main challenges and opportunities during their early careers. Crucial points are networking, guidance, mentoring, as well as funding acquisition and the personal work-life balance.
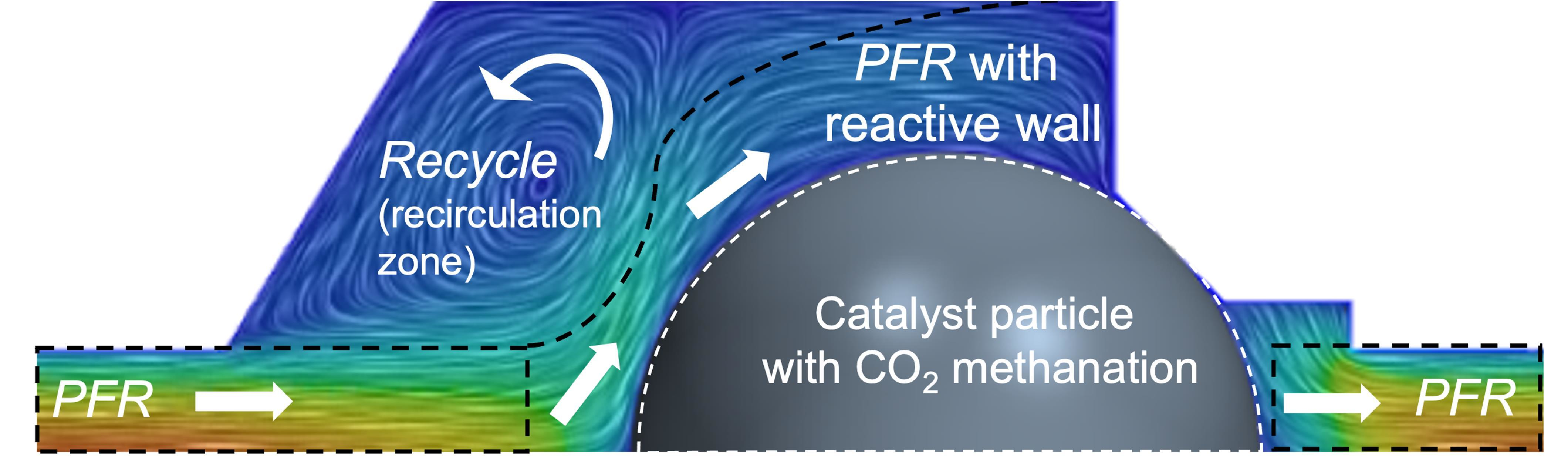
Catalysts
· 2022
The Temkin reactor can be applied for industrial relevant catalyst testing with unmodified catalyst particles. It was assumed in the literature that this reactor behaves as a cascade of continuously stirred tank reactors (CSTR). However, this assumption was based only on outlet gas composition or inert residence time distribution measurements. The present work theoretically investigates the catalytic CO2 methanation as a test case on different catalyst geometries, a sphere, and a ring, inside a single Temkin reaction chamber under isothermal conditions. Axial gas-phase species profiles from detailed computational fluid dynamics (CFD) are compared with a CSTR and 1D plug-flow reactor (PFR) model using a sophisticated microkinetic model. In addition, a 1D chemical reactor network (CRN) model was developed, and model parameters were adjusted based on the CFD simulations. Whereas the ideal reactor models overpredict the axial product concentrations, the CRN model results agree well with the CFD simulations, especially under low to medium flow rates. This study shows that complex flow patterns greatly influence species fields inside the Temkin reactor. Although residence time measurements suggest CSTR-like behavior, the reactive flow cannot be described by either a CSTR or PFR model but with the developed CRN model.
Nachrichten aus der Chemie
· 2022
Von atomaren Prozessen bis zu Stoffkreisläufen – die technische Chemie arbeitet auf allen Skalen am Ziel, die CO2-Emissionen zu senken. Dafür werden Kreislaufwirtschaften eingerichtet, Reaktoren werden mit „grünem“ Strom beheizt, und die Multiskalen-Modellierung sucht nach effizienteren Katalysatoren.
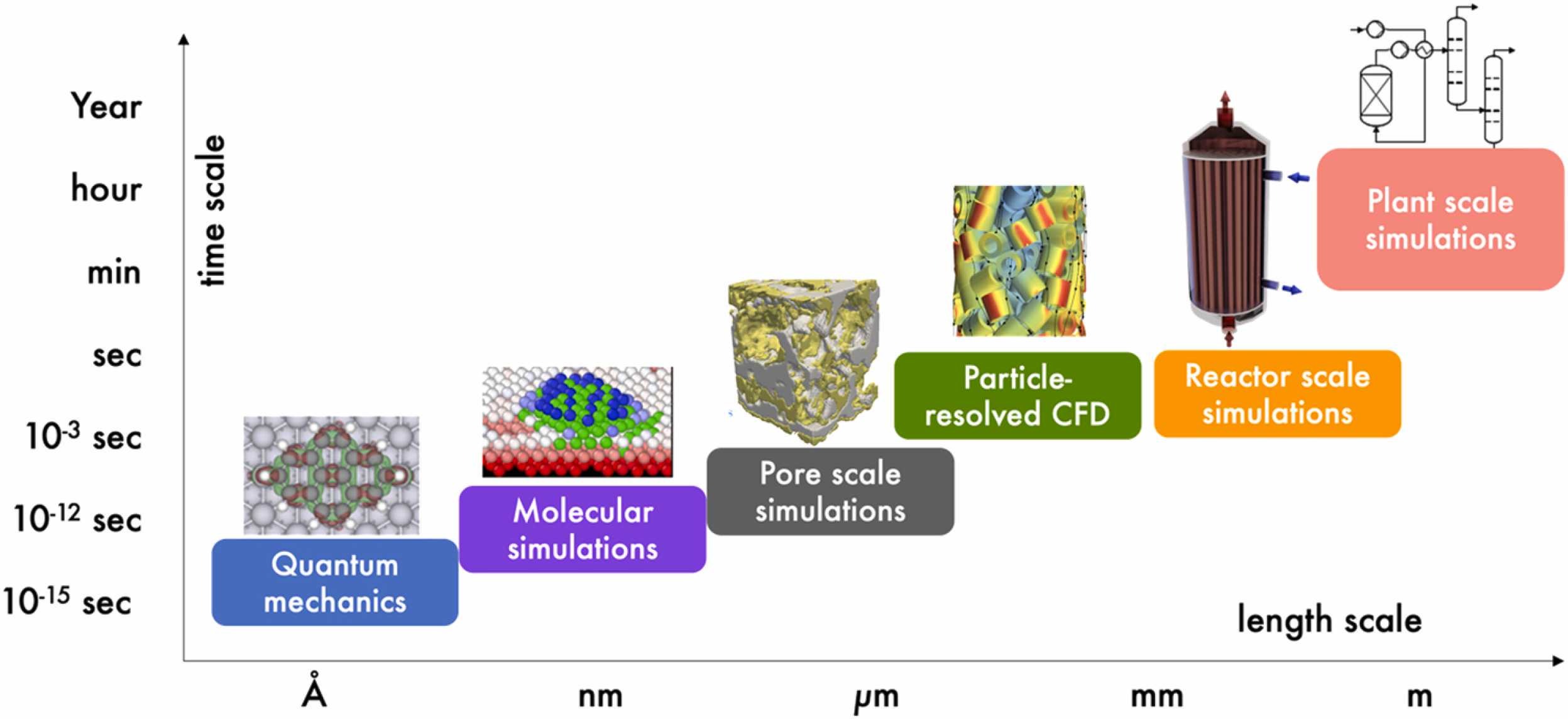
Chemical Engineering Research and Design
· 2022
This work reports the results of a perspective workshop held in summer 2021 discussing the current status and future needs for multiscale modeling in reaction engineering. This research topic is one of the most challenging and likewise most interdisciplinary in the chemical engineering community, today. Although it is progressing fast in terms of methods development, it is only slowly applied by most reaction engineers. Therefore, this perspective is aimed to promote this field and facilitate research and a common understanding. It involves the following areas: (1) reactors and cells with surface changes focusing on Density Functional Theory and Monte-Carlo simulations; (2) hierarchically-based microkinetic analysis of heterogeneous catalytic processes including structure sensitivity, microkinetic mechanism development, and parameter estimation; (3) coupling first-principles kinetic models and CFD simulations of catalytic reactors covering chemistry acceleration strategies and surrogate models; and finally (4) catalyst-reactor-plant systems with details on linking CFD with plant simulations, respectively. It therefore highlights recent achievements, challenges, and future needs for fueling this urgent research topic in reaction engineering.
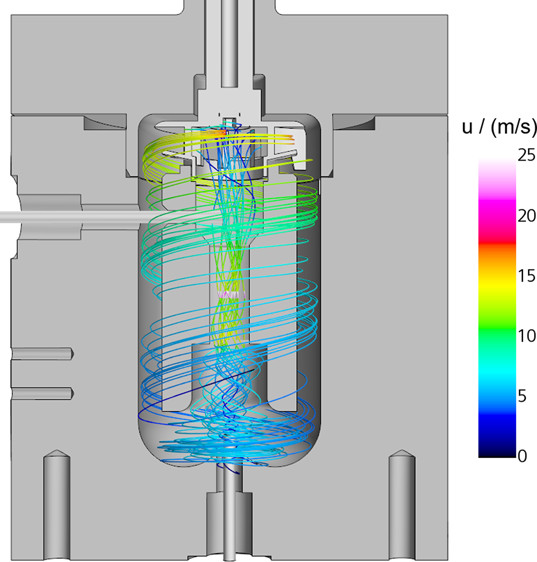
Industrial & Engineering Chemistry Research
· 2022
The Berty reactor is a continuously operated stirred tank reactor that is used in catalytic experiments to achieve gradientless conditions. Uniformity of the concentration profile has been confirmed through residence time measurements, but the temperature profiles originating from the heat of reaction have not been investigated yet. In this work, the profiles inside the reactor are investigated for the exothermic methanation of CO2 under typical operating conditions for kinetic measurements via Computational Fluid Dynamics simulations. The results indicate that temperature gradients in the vicinity of the catalyst can occur even though residence time measurements confirm uniformity of concentration. It was shown that a low conversion per pass is key to uniform conditions, which can be influenced by a high dilution ratio of the catalyst bed, high operating pressure, and high rotation speed.
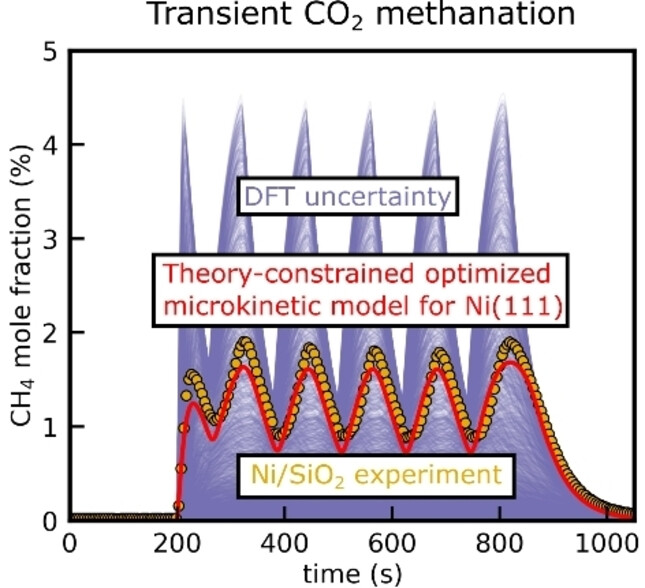
ChemCatChem
· 2022
The transient operation of methanation reactors can become desirable when coupled with fluctuating renewable energies in Power-to-Gas scenarios, which requires suitable kinetic approaches that can describe the transient catalytic phenomena. A combined experimental and theoretical investigation of the transient CO2 methanation is conducted using concentration forcing to derive a suitable microkinetic model. Methanation experiments are performed with a Ni/SiO2 catalyst in a Berty-type reactor at industrially relevant conditions. The microkinetics are based on previous work and were automatically constructed for the Ni(111) facet using the Reaction Mechanism Generator. A feasible set of energetic parameters of the microkinetic models was identified in a theory-constrained optimization procedure within the DFT uncertainty space that can accurately reproduce the experimental results on a first-principles basis. The microkinetic model unravels that the formation of H2O* and CH3* control the activity and selectivity of Ni(111) under the investigated conditions.
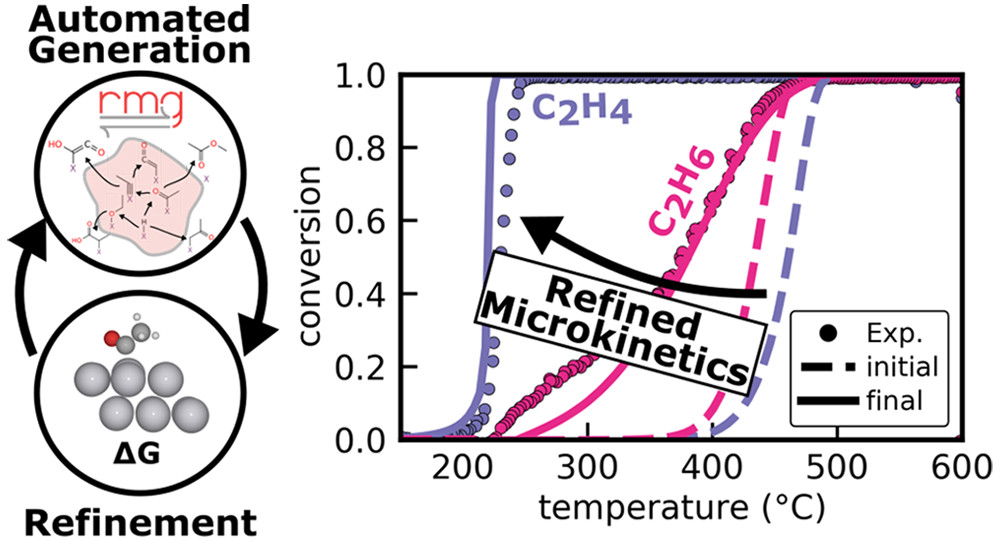
ACS Catalysis
· 2022
Emissions from vehicles contain a variety of pollutants that must be either oxidized or reduced efficiently in the catalytic converter. Improvements to the catalyst require knowledge of the microkinetics, but the complexity of the exhaust gas mixture makes it challenging to identify the reaction network. This complexity was tackled by using the “Reaction Mechanism Generator” (RMG) to automatically generate microkinetic models for the oxidation of combustion byproducts from stoichiometric gasoline direct injection engines on Pt(111). The possibilities and the limitations encountered during the generation procedure are discussed in detail. A combination of first-principles-based mechanism construction and top-down parameter refinement allows a description of experimental results obtained by kinetic testing of a Pt/Al2O3 monolith under stoichiometric conditions. The study can serve as a blueprint for the usage of RMG for other challenging heterogeneously catalyzed reactions.
Chemie Ingenieur Technik
· 2021
Im vorliegenden Artikel werden die Ergebnisse des NaWuReT-Workshops (Nachwuchs Reaktionstechnik) aus dem Dezember 2020 zum Thema „Machen wir relevante Wissenschaft?“ vorgestellt. Nach Einführungs- und Übersichtsvorträgen wurden die Themen „(Un)überwindbare Hürden für Citizen Scientists in der Reaktionstechnik?“ und „Circular Economy in der Reaktionstechnik mit/für die Gesellschaft?“ diskutiert und vielfältige Ideen und Anregungen aus der Diskussion abgeleitet.
PhD Thesis, TU Clausthal
· 2021
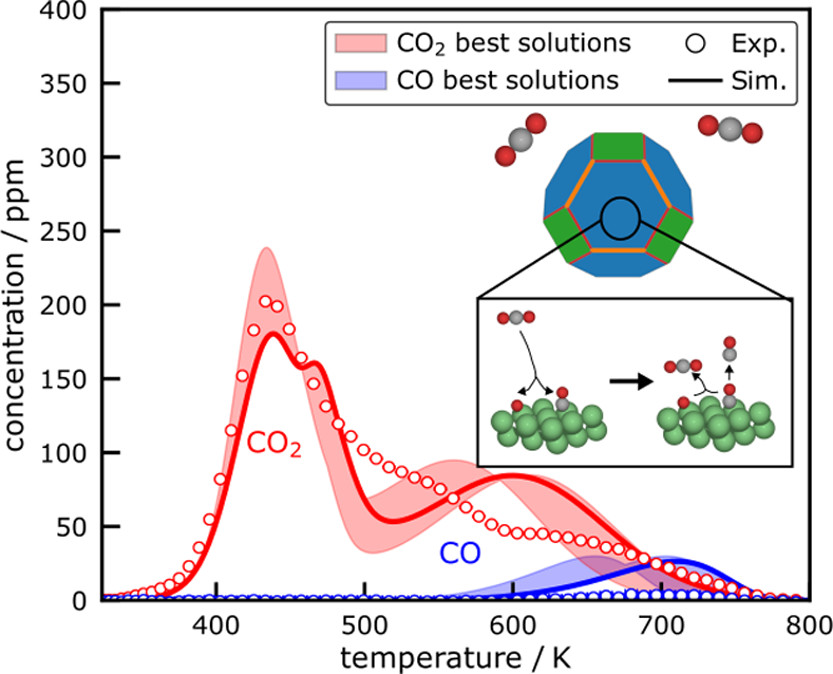
Journal of Physical Chemistry C
· 2021
The conversion of CO2 into valuable products with hydrogenation reactions on Ni catalysts requires a fundamental understanding of the interaction of CO2 with the Ni surface. Microkinetic modeling is used to investigate the interaction of CO2 with a multifaceted Ni crystal and compared to temperature-programmed desorption (TPD) experiments from supported Ni catalysts. TPD experiments were performed for Ni/γ-Al2O3 and Ni/SiO2 catalysts. Uncertainties in the DFT-derived model parameters were accounted for by a global uncertainty analysis (GUA). On the basis of the model, the adsorption and desorption of CO2 exhibit a structure–sensitivity for the investigated Ni facets. Whereas the initial multifaceted TPD model is not in quantitative agreement with the experimentally recorded desorption profile, the GUA reveals a feasible set of model parameters that are in close agreement with the data. The comparison of the desorption profiles of the different catalysts and the multifacet simulation shows that desorption of CO2 from basic sites and desorption from the Ni facets overlap and thus have similar desorption kinetics.
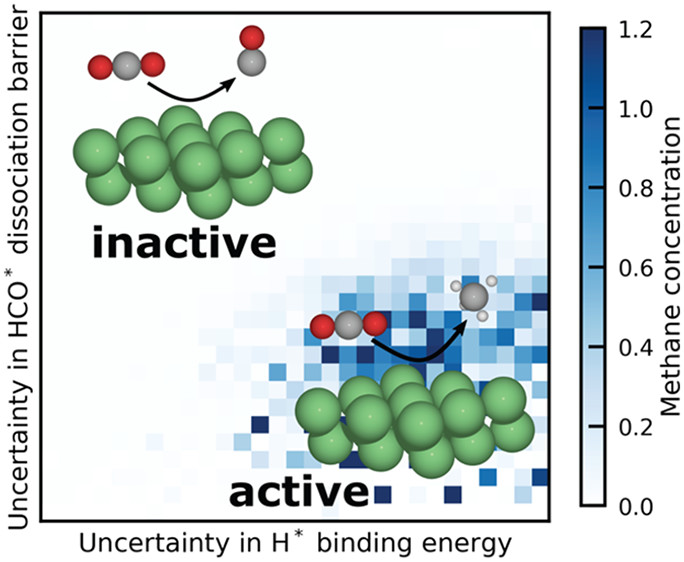
JACS Au
· 2021
Automatic mechanism generation is used to determine mechanisms for the CO2 hydrogenation on Ni(111) in a two-stage process while considering the correlated uncertainty in DFT-based energetic parameters systematically. In a coarse stage, all the possible chemistry is explored with gas-phase products down to the ppb level, while a refined stage discovers the core methanation submechanism. Five thousand unique mechanisms were generated, which contain minor perturbations in all parameters. Global uncertainty assessment, global sensitivity analysis, and degree of rate control analysis are performed to study the effect of this parametric uncertainty on the microkinetic model predictions. Comparison of the model predictions with experimental data on a Ni/SiO2 catalyst find a feasible set of microkinetic mechanisms within the correlated uncertainty space that are in quantitative agreement with the measured data, without relying on explicit parameter optimization. Global uncertainty and sensitivity analyses provide tools to determine the pathways and key factors that control the methanation activity within the parameter space. Together, these methods reveal that the degree of rate control approach can be misleading if parametric uncertainty is not considered. The procedure of considering uncertainties in the automated mechanism generation is not unique to CO2 methanation and can be easily extended to other challenging heterogeneously catalyzed reactions.
Computer Aided Chemical Engineering (ESCAPE 30)
· 2020
In this contribution, we present a detailed three-dimensional computational fluid dynamics (CFD) model of a fixed-bed reactor processing the catalytic CO2 methanation reaction. Due to the small tube-to-particle-diameter ratio, particle-resolved CFD simulations are carried out, since they can locally account for transport phenomena, i.e. momentum, heat, and mass transfer. In addition to the interstitial flow simulation, three-dimensional diffusion plus reaction is modelled inside the catalytic pellets. The comparison in terms of axial temperature with a one-dimensional model shows the discrepancy between the models. Additional CFD simulations with different pellet shapes show that the shape has little influence on the heat transfer in these slender fixed-bed arrangements. Experimental results of axial concentration profiles will further validate the CFD model.
Computer Aided Chemical Engineering (ESCAPE 30)
· 2020
The automated reaction mechanism generator (RMG) is used to investigate the methanation of CO2 on the Ni(111) and Ni(211) surface. Linear scaling relations are applied for the thermochemistry of the adsorbates, which are compared to state-of-the-art electronic structure calculations and show a reasonable predictability. RMG discovers nearly the same amount of species and reactions for both facets. However, a reaction path analysis in a reactor simulation shows that the reaction pathways on both surfaces differ significantly, which is caused by the difference in the binding energy of the adsorbates. Reactor simulations reveal a lower methane production rate compared to experiments obtained in a Berty reactor with a Ni/Al2O3 catalyst, which is a result of a high surface coverage with CO⁎ and demands, therefore, the inclusion of a coverage dependent heat of formation of the adsorbates.
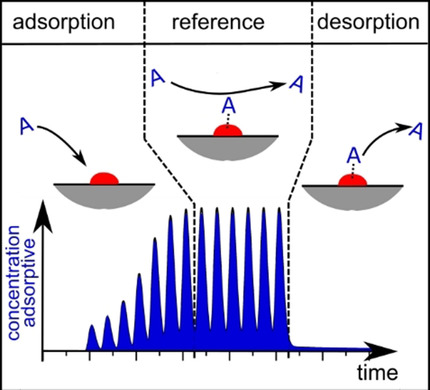
ChemCatChem
· 2020
An improved pulse-chemisorption technique is presented, which is proven to be independent of the strength of interaction between adsorptive and catalyst material. The methodology is based on the transient mass balance of the adsorptive, allowing for quantitative evaluation of the pulse-signal obtained from the experiment. Two experimental strategies for determination of the adsorption capacity are introduced, with and without using an internal standard. The methodology is illustratively discussed based on simulation results and verified by chemisorption experiments of hydrogen and carbon dioxide on a supported nickel catalyst. In addition, temperature and dosing effects are examined and benchmarked with volumetric measurements of the adsorption capacity. The proposed mathematical evaluation method of the measured data can be applied directly to experiments performed in standard equipment for pulse-chemisorption or in modified catalyst test rigs to measure adsorption capacity before and after reaction experiments. The technique shows potential for determination of sorption kinetics and combination with operando spectroscopy.
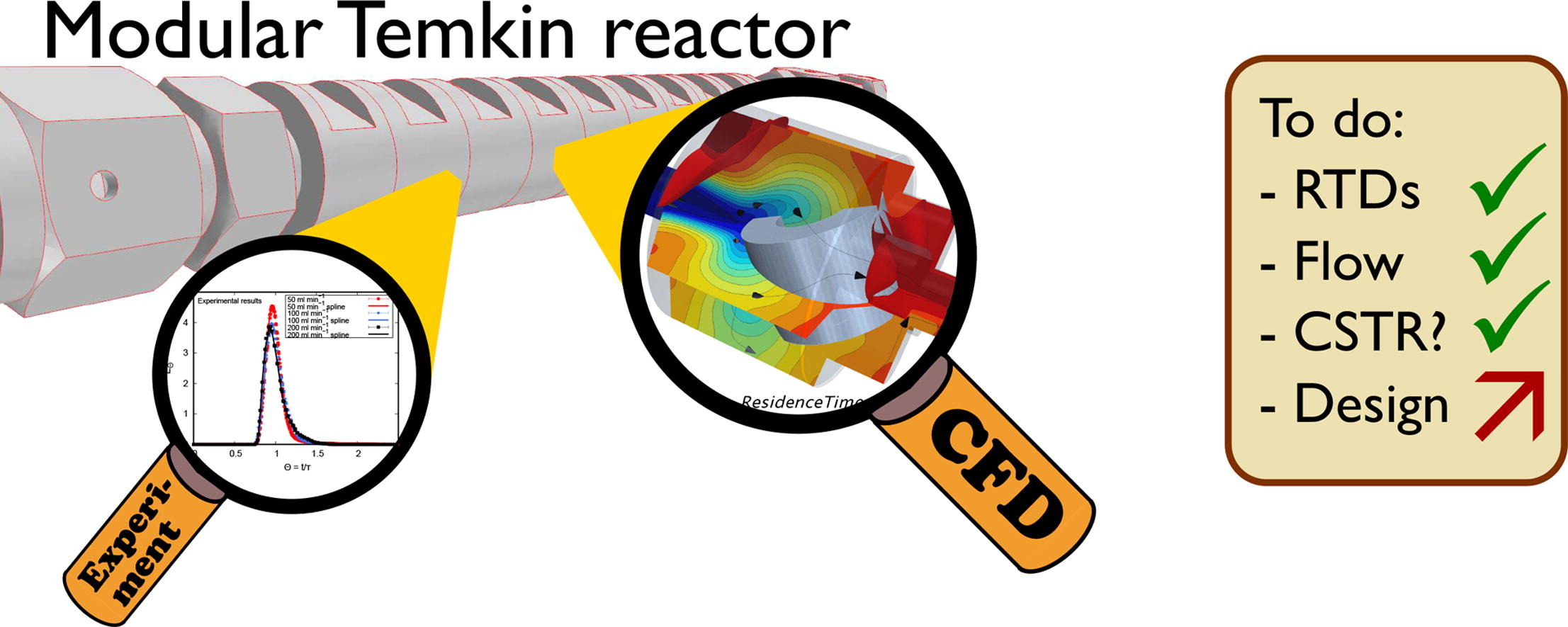
Chemical Engineering Journal
· 2020
The determination of reaction kinetics without the influence of transport limitations is challenging, especially under industrially relevant conditions, particle shapes, and sizes. The Temkin reactor concept, developed in the 1960s, allows to measure reaction kinetics with a small number of unmodified catalyst particles operating under assumed isothermal and plug-flow conditions. In this study, a recently developed modular Temkin reactor design is studied experimentally and with detailed CFD simulations in order to shed light on the residence time distribution without reaction. Cold fluid volumetric flow rates are varied in the range of 50–200 and two different particle shapes (spheres and rings) are tested. Pulse experiments and passive scalar computational fluid dynamics (CFD) simulations verify that this reactor design behaves like a cascade of stirred tank reactors with Bodenstein numbers of each chamber smaller than three. Mean age of air CFD simulations highlight potentials to re-design the entrance of the chamber in order to minimize stagnation zones.
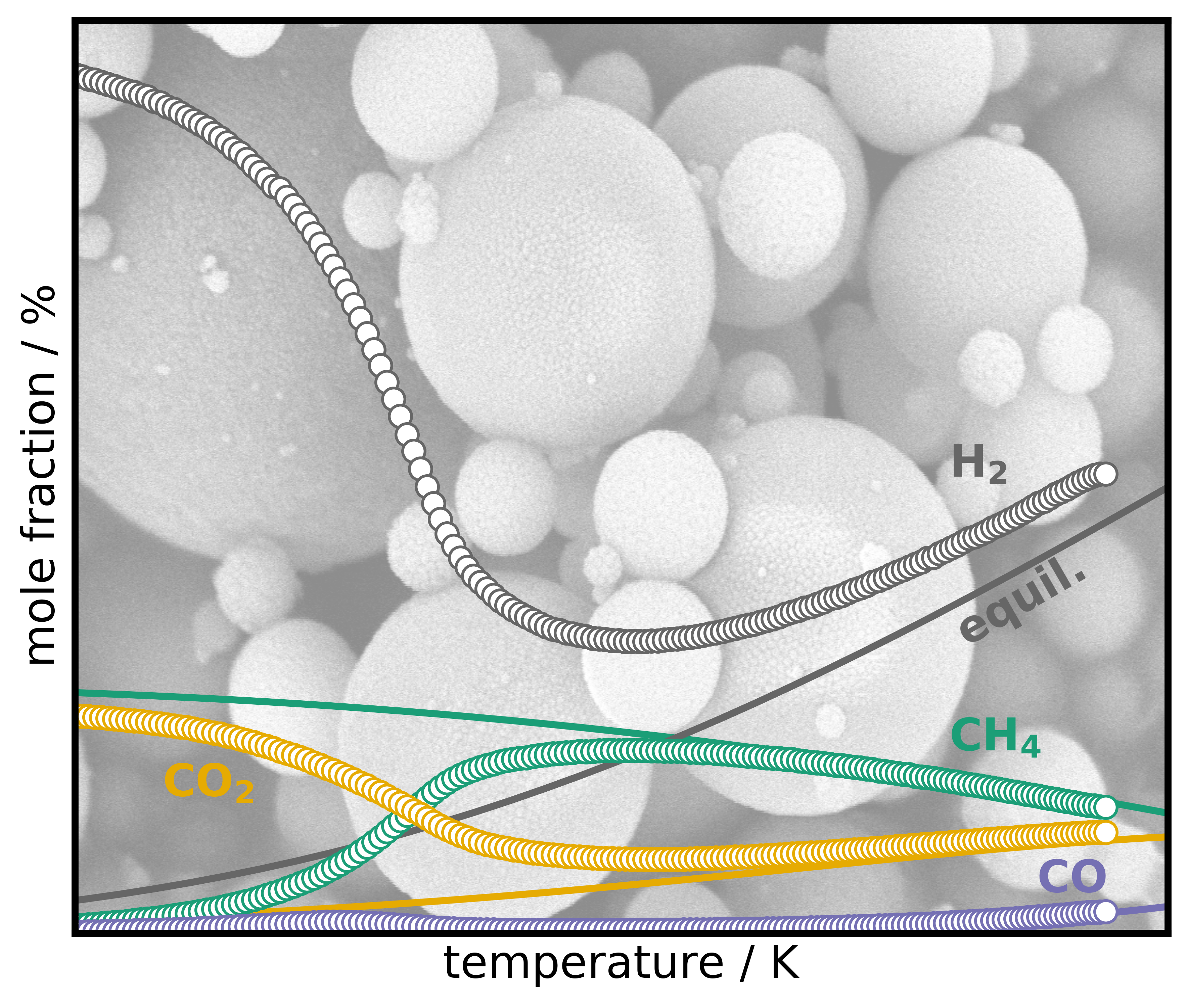
Catalysts
· 2020
A catalyst production method that enables the independent tailoring of the structural properties of the catalyst, such as pore size, metal particle size, metal loading or surface area, allows to increase the efficiency of a catalytic process. Such tailoring can help to make the valorization of CO2 into synthetic fuels on Ni catalysts competitive to conventional fossil fuel production. In this work, a new spray-drying method was used to produce Ni catalysts supported on SiO2 and Al2O3 nanoparticles with tunable properties. The influence of the primary particle size of the support, different metal loadings, and heat treatments were applied to investigate the potential to tailor the properties of catalysts. The catalysts were examined with physical and chemical characterization methods, including X-ray diffraction, temperature-programmed reduction, and chemisorption. A temperature-scanning technique was applied to screen the catalysts for CO2 methanation. With the spray-drying method presented here, well-organized porous spherical nanoparticles of highly dispersed NiO nanoparticles supported on silica with tunable properties were produced and characterized. Moreover, the pore size, metal particle size, and metal loading can be controlled independently, which allows to produce catalyst particles with the desired properties. Ni/SiO2 catalysts with surface areas of up to 40 m2 g−1 with Ni crystals in the range of 4 nm were produced, which exhibited a high activity for the CO2 methanation.
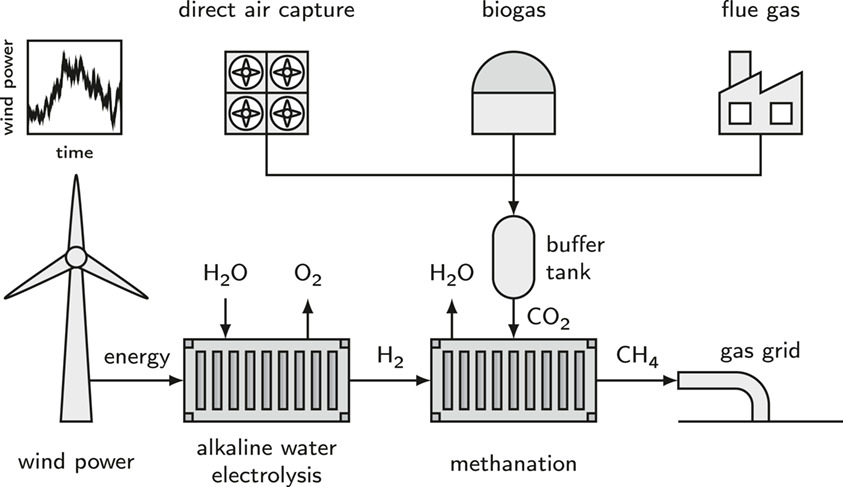
Chemie Ingenieur Technik
· 2020
The dynamic operation of a power-to-gas plant powered by wind energy is theoretically studied by coupling an empirical model of an alkaline water electrolyzer with a 1D heterogeneous model of a methanation reactor. H2 produced by the electrolyzer follows the wind power profile, but operation in the part-load range can raise safety concerns. The dynamically generated methane quality comes close to the required value for injection into the gas grid, if the stoichiometric ratio is controlled. To satisfy the gas quality at all times, it is necessary to design a more tolerant reactor.
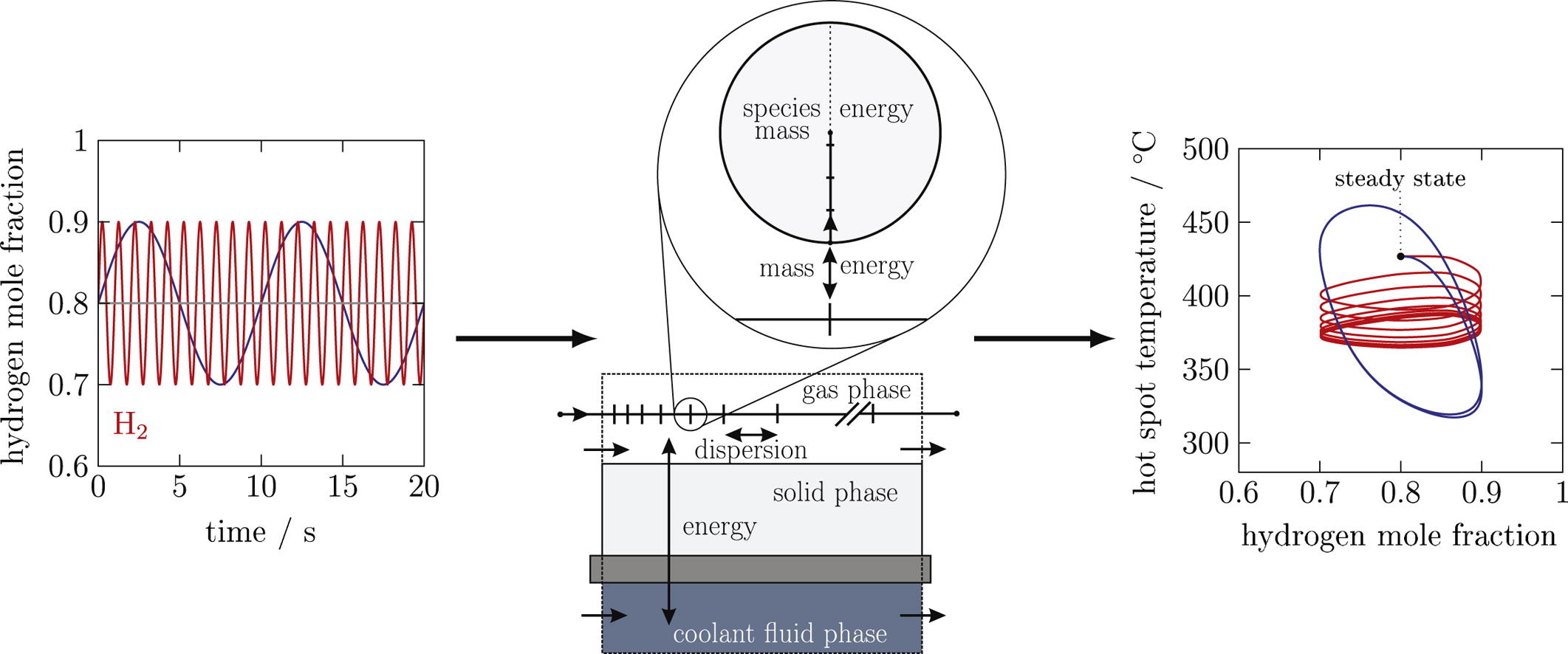
Chemical Engineering Science
· 2019
The dynamic operation of methanation reactors is under discussion due to the increasing interest in power-to-gas technology. Here, hydrogen is supplied via a water electrolyzer which is powered by fluctuating renewable energy sources. With dynamic process modeling and simulation, the methanation reactor can be designed. Since the methanation reaction proceeds fast in the presence of an active catalyst and releases a large amount of reaction heat, the model has to account for the pellet phase in terms of energy, species mass and reaction. In this work, a dynamic 1D heterogeneous model is presented. The model describes a micro-structured fixed-bed reactor for the methanation of carbon dioxide under industrially relevant operating conditions with periodic oscillations of the inlet feed composition. The simulation shows that the hot spot temperature changes significantly during the periodic operation, both in position and magnitude. For high frequency changes the reactor behavior moves towards a thermally relaxed steady state. The outlet methane concentration in the dynamic operation does not reach the steady-state value. However, the model reveals interesting insights of the dynamic CO2 methanation, which will be accompanied with experiments in the future.
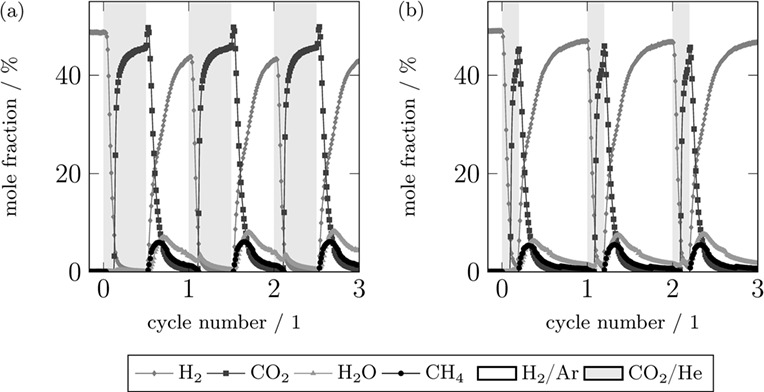
Chemie Ingenieur Technik
· 2019
Investigating the effect of concentration forcing of the CO2 methanation is not only relevant for power-to-gas plants but also for the study of dynamic phenomena of this reaction. In this study a Ni/Al2O3 catalyst is investigated under concentration forcing at industrially relevant conditions. The dynamic experiments allow an evaluation in terms of the reaction rate and enable the study of the reaction mechanism. The experiments show that no methane is formed in the CO2-rich part of the cycle, whereas a fast hydrogenation of carbonaceous species to methane takes place upon switching to H2.
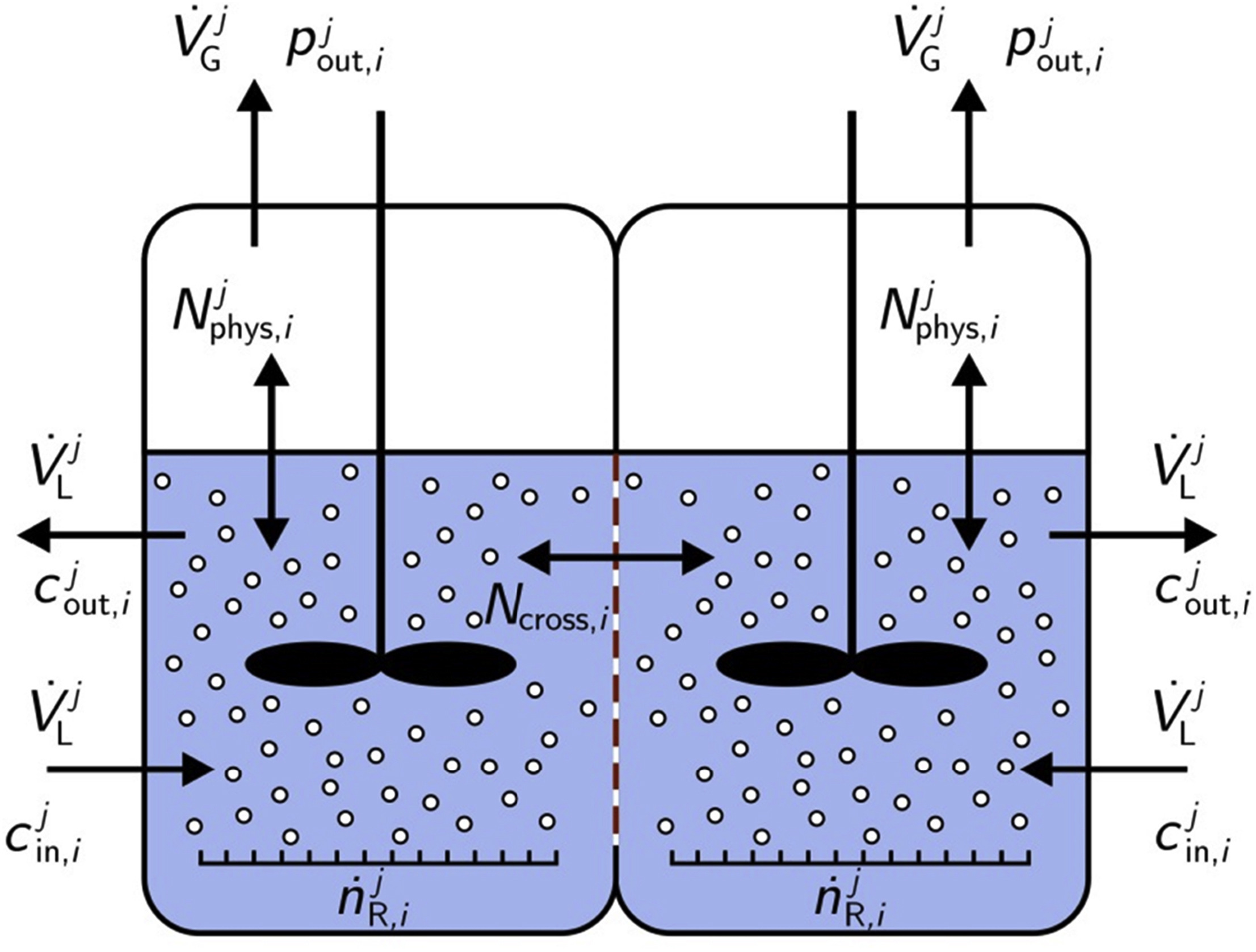
International Journal of Hydrogen Energy
· 2017
In this paper a model for the prediction of the product gas purity in alkaline water electrolysis is proposed. For the estimation of the exhaust gas compositions the operating conditions, such as current density, electrolyte flow rate, concentration and temperature as well as process management possibilities are considered. The development of the model relies on a classical process engineering approach and depicts the electrolysis cell through coupled continuously stirred-tank reactors. Furthermore, the mass transport phenomena between the phases are considered through the application of Reynolds and Sherwood correlations. Finally, the validation of the model is performed through experiments, which are carried out in a lab-scale electrolyzer with a 150 cm2 zero-gap cell and KOH electrolyte at atmospheric pressure. This investigation reveals that gas purity in alkaline water electrolysis is mainly affected by mixing the anodic and cathodic electrolyte cycles, which transport dissolved electrolysis products into the opposite half cell compartments. However, this transport mechanism can be significantly reduced by adjustment of the operating conditions of the electrolyzer.
Publication list auto-updated via Semantic Scholar and arXiv. Last updated: July 2026.