Research
The Kreitz group studies the detailed chemical kinetics of complex heterogeneously catalyzed reactions, including the upcycling of plastic waste, hydroformylation, fuel synthesis, and mechanocatalysis. We combine electronic structure theory, automated mechanism generation, multiscale modeling, and machine learning with kinetic experiments to build predictive models of catalytic reactions — from the elementary surface reactions to the reactor scale.
Automated Mechanism Generation
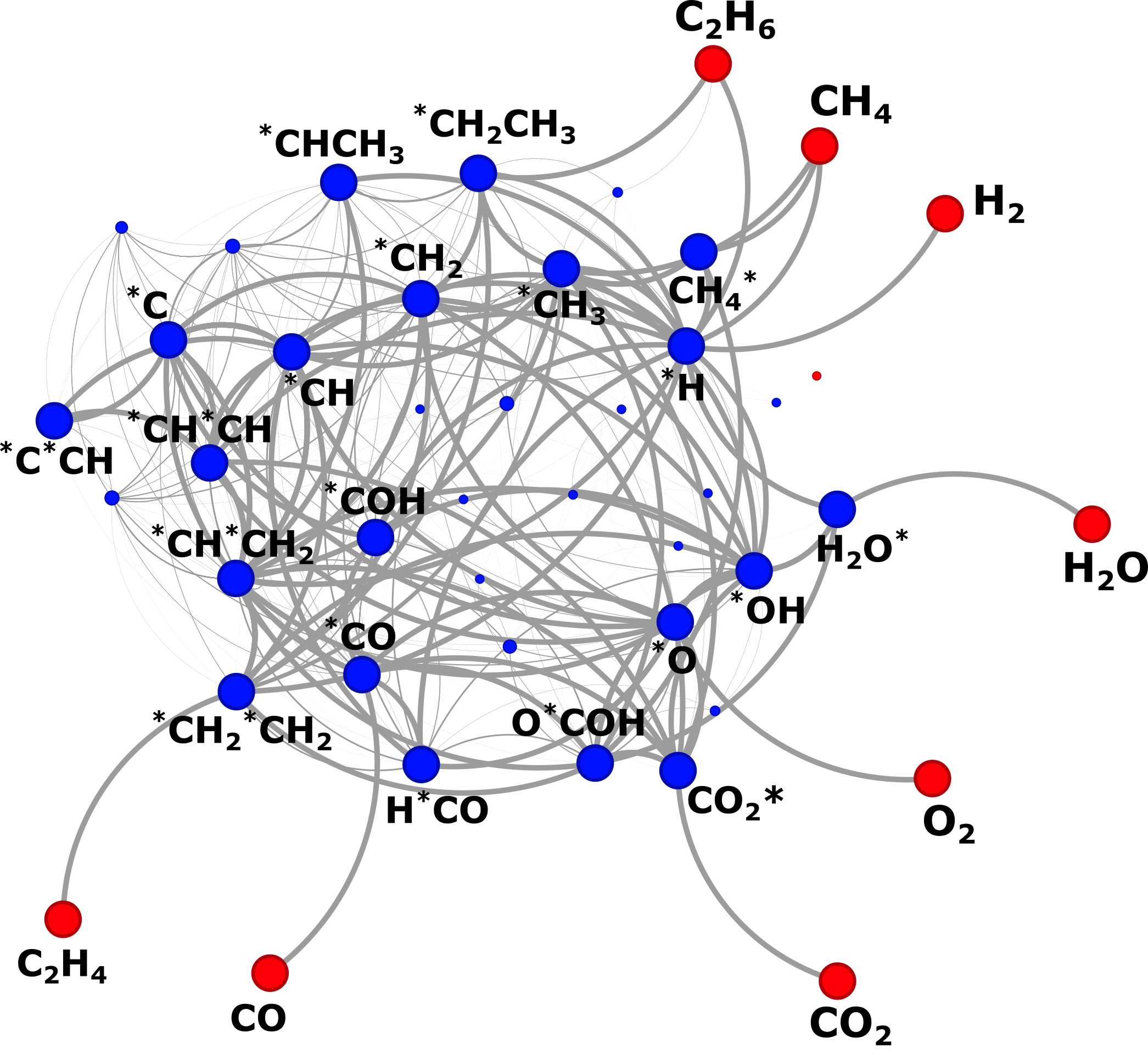
Heterogeneous catalytic reactions involve thousands of elementary steps across complex catalyst surfaces, making manual mechanism construction impractical. The group develops automated mechanism generation methods and contributes to the open-source Reaction Mechanism Generator (RMG) to accelerate the development of predictive microkinetic models. Current efforts focus on extending these approaches to metal oxides, multifaceted nanoparticles, and larger reactants.
A central challenge is the uncertainty in DFT-derived energetic parameters, which propagates to reaction mechanisms and kinetic predictions. By combining RMG with correlated uncertainty quantification, we generate ensembles of reaction mechanisms to identify pathways and parameter sets consistent with experiments without ad hoc fitting, enabling a direct connection between first-principles calculations and experimentally observable kinetics.
Representative work:
- Mechanism generation with correlated uncertainty quantification for emission oxidation catalysis (Angewandte Chemie 2023)
- Automated mechanism generation for CO₂ methanation on Ni(111) with uncertainty quantification (JACS Au 2021)
- Automated catalysts screening and multidentate adsorbates in RMG (Digital Discovery 2024)
Multiscale Modeling of Catalytic Reactions
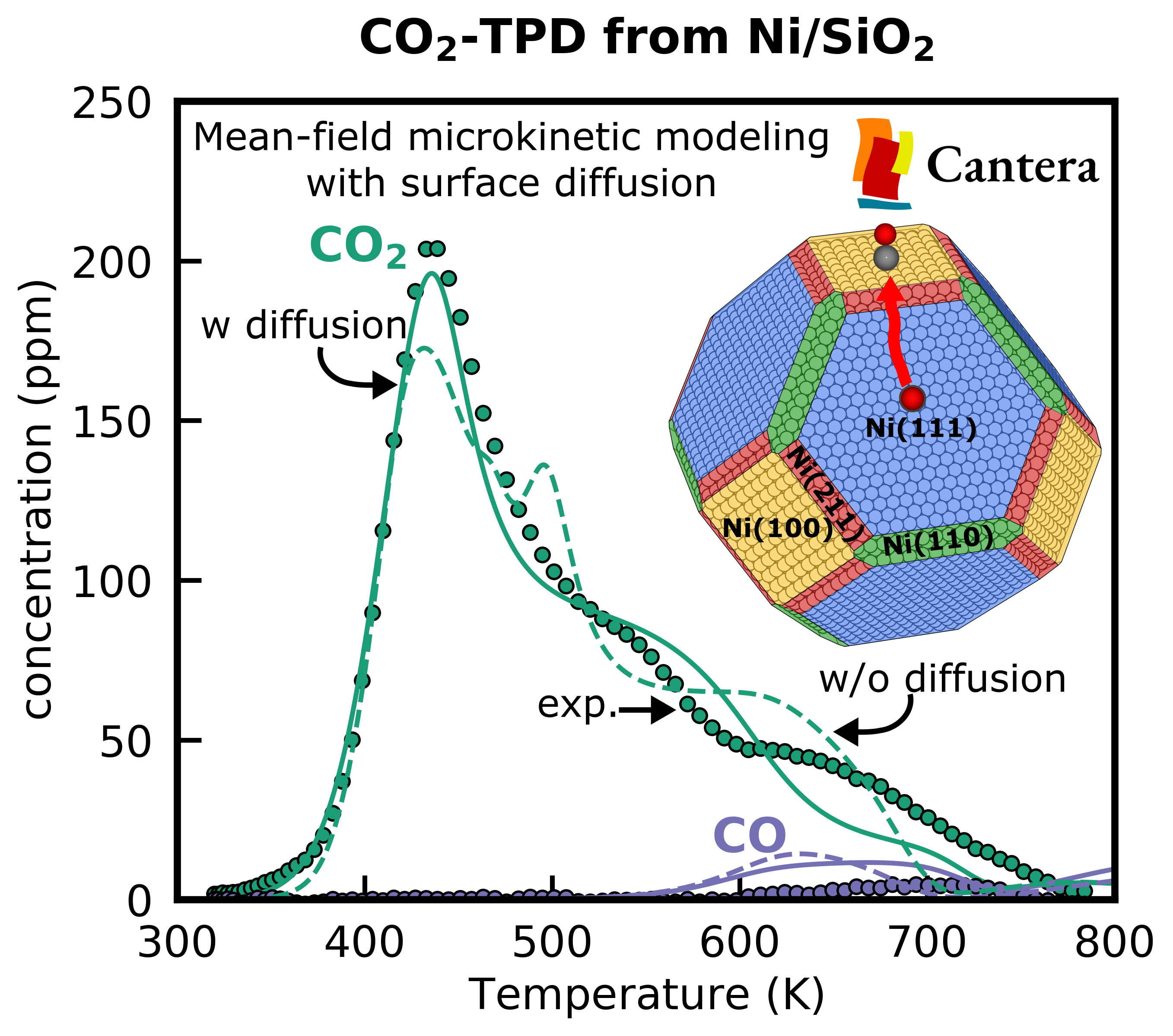
Detailed reaction mechanisms must ultimately reproduce experimentally observable reactor behavior. The group develops first-principles-based multiscale models that bridge atomistic simulations and reactor-scale performance across various length and time scales. A major focus is overcoming the material gap between idealized single-crystal facet models and technical catalysts composed of supported metal nanoparticles.
We develop structure-dependent models for multifaceted nanoparticles that account for the contributions of different surface facets and diffusion of adsorbates. Our main software for the multiscale simulation is the open-source Cantera toolkit. Additional efforts focus on incorporating coverage effects and accelerating reactor-scale simulations through surrogate models that enable the integration of detailed microkinetics into CFD and reactor models.
Representative work:
- Structure-dependent microkinetics with surface diffusion in Cantera (Journal of Catalysis 2025)
- Automated microkinetics for exhaust gas oxidation over Pt with DFT-constrained optimization (ACS Catalysis 2022)
- Multilevel on-the-fly sparse grids for coupling microkinetics with reactor simulations (Computers & Chemical Engineering 2025)
- Coverage-dependent thermophysical properties in Cantera (JCIM 2025)
Thermochemistry of Adsorbates

Predictive microkinetic models require accurate thermodynamic and kinetic parameters that define the catalytic free-energy landscape. However, enthalpies of formation of adsorbates derived from DFT contain large uncertainties that limit predictive modeling.
The group develops thermochemical methods based on error cancellation that preserve the bonding environment and hybridization of adsorbates through carefully chosen reference reactions. These approaches enable DFT-derived enthalpies of formation with experimental accuracy across different exchange–correlation functionals and provide a link between ab initio calculations, experimental surface science data, and gas-phase thermochemistry.
Additional efforts focus on standardizing thermochemical conventions in computational catalysis and incorporating coverage-dependent thermophysical properties into Cantera to capture adsorbate–adsorbate interactions at realistic surface coverages.
Representative work:
- Unified framework for thermochemistry concepts in computational heterogeneous catalysis (Chemical Society Reviews 2025)
- Generalized thermochemical hierarchy linking experimental and ab initio adsorbate enthalpies (JCTC 2023)
- CBH-based enthalpies on Pt(111), Ni(111), and MgO(100) (Faraday Discussions 2026)
Machine Learning for Catalysis
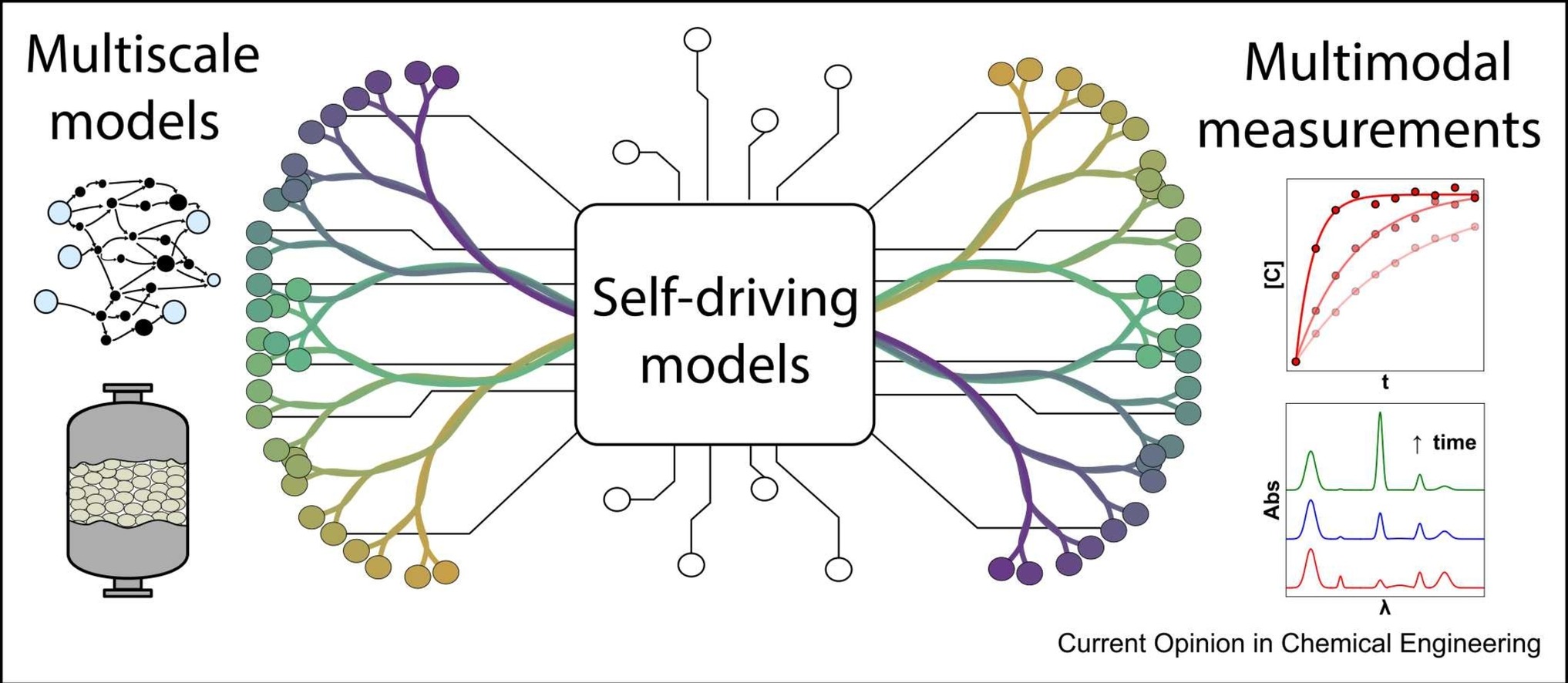
The group develops and applies machine learning methods to accelerate mechanism generation, atomistic simulations, and multiscale modeling workflows for heterogeneous catalysis. A major focus is the integration of generative and agentic AI with first-principles modeling to enable autonomous exploration of catalytic reaction networks and catalyst design spaces.
Representative work:
- Prospects for AI in understanding intrinsic kinetics of heterogeneous catalysis (Current Opinion in Chemical Engineering 2026)
For a full list of publications see the Publications page.